
Disease Modeling
Research Group

Our research group is committed to identifying the genetic causes and developing potential cures for genetic disorders.
We apply cutting edge sequencing technologies to identify the causes of rare diseases within the genome of affected individuals. To understand the impact of such genetic changes, we develop disease models to gain deep insights into the underlying pathomechanisms. This approach enables us to uncover the genetic origins and path the way to personalized treatments of neurodevelopmental disorders and beyond.”
Prof. Dr. med. Nataliya Di Donato
Group leader, Director Department for Human Genetics
Publications
on PubMed
Research Focus
Uncovering Hidden Genetic Alterations in Rare and Complex Diseases
Rare diseases are very difficult to diagnose as they are very diverse and can range from increased infection susceptibility to neurological impairment. Although each of the conditions affects less than 0.05% of the general population in the EU, it is estimated that a total of 27 to 36 million people suffer from a rare disease in Europe. Typically, rare diseases manifest early in life, sending children and their parents on a diagnostic odyssey for years. Most rare diseases are caused by genetic alterations, making genetic testing the most promising tool for a fast and accurate diagnosis.
The integrated use of third-generation sequencing techniques allows us to gain insights into the interplay between genetic variations and phenotypic outcomes. Leveraging superior (epi-)genomic and transcriptomic sequencing technologies helps us understand rare diseases and opens up the possibility to explore fundamental biological mechanisms and their contribution to disease manifestation.
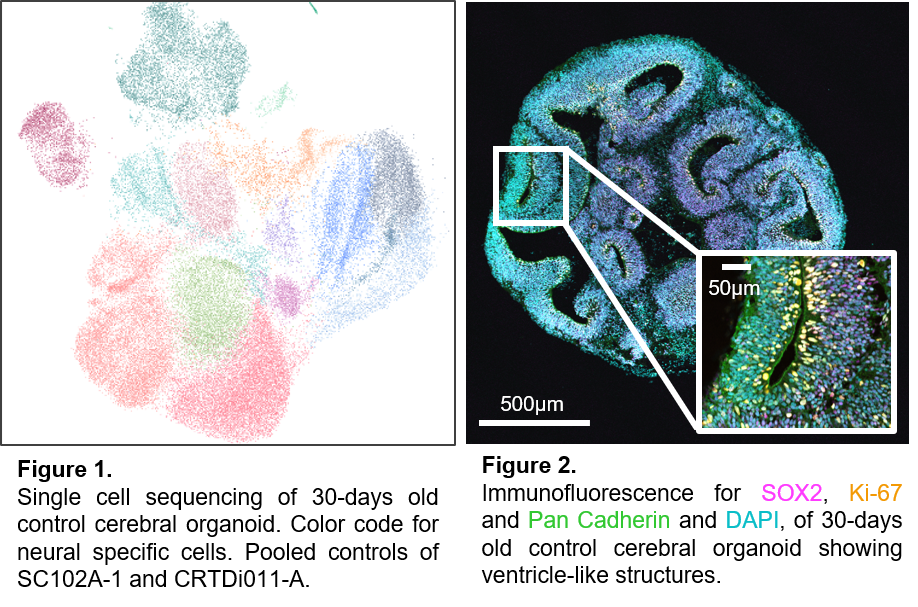
Understanding genetic variants remains a major challenge in human genetics. Although advanced sequencing technologies allow us to identify even complex genomic variants, it is difficult to classify these variants as benign or pathogenic due to limited evidence. About half of all genetic variants fall into the category “variants of uncertain significance” (VUS), which means diagnostic uncertainty for patients and their families.
In other instances, such as non-muscle actinopathies, genetic alterations are associated with a wide range of clinical manifestations. Although patients are diagnosed with the same disorder, only a subset exhibit specific symptoms. The underlying reasons for the variability in clinical penetrance remain unclear.
Therefore, our research focuses on the functional analysis of rare genetic variants. By studying the effects of these variants in in vivo disease model systems, such as patient-derived cells and organoids, we aim to uncover the mechanisms that drive disease phenotypes. This functional research provides valuable insights that link genetic mutations to molecular pathways, helping to develop targeted therapeutic strategies for individuals with (rare) genetic disorders.

Many genetic disorders are associated with neurodevelopmental impairment. Our research is dedicated to uncovering the genetic foundations of these neurological changes to ultimately support tailored therapeutic approaches. Our interdisciplinary approach of medical expertise with direct connection to patients and scientists allows us to trace genetic mutations from a patient's symptoms down to the specific molecular mechanisms involved.
Mutations in the actin genes ACTB and ACTG1 can result in developmental disorders and organ malformations collectively referred to as non-muscular actinopathies (NMA). Nevertheless, the full mutational and clinical spectrum of human monogenic disorders caused by ACTB or ACTG1 variants has yet to be systematically explored. Challenges in assigning pathogenicity and in linking individual variants to specific NMAs hamper accurate diagnosis. Even after a specific NMA is diagnosed, clinical management and providing prognosis is challenging due to limited data.
Our group aims to uncover the mechanisms underlying NMA and explore the connections between different genetic mutations and their associated disease phenotypes. Our NMA patient registry is a valuable resource for understanding the varied outcomes of genetic variants, exploring their molecular effects, and uncovering the mechanisms behind pathogenicity. Studying these rare conditions not only provides insights into the function of one of the most essential proteins but also paves the way for the potential development of targeted therapies for individuals affected by rare actinopathies and the associated disorders, like Baraitser-Winter syndrome, Dystonia-Deafness syndrome and others.
We are continuously working on growing our patient database. If you are interested in joining our research please get in contact.
Downloads ACTB / ACTG Patient Registry
- Study information ACTB / ACTG patients (EN / DE)
- Informed consent ACTB / ACTG patients (EN / DE)
- Study information ACTB / ACTG legal guardians (DE)
- Informed consent ACTB / ACTG legal guardians (DE)
- Study information parents (EN / DE)
- Informed consent parents (EN / DE)

