
More news on the homepage
In vivo gene correction in hereditary haemochromatosis
With a prevalence of 0.2 - 0.4%, congenital haemochromatosis type 1 is one of the most common congenital metabolic diseases in Europe.
In over 80% of cases, a homozygous C282Y mutation is present in the HFE gene, which causes misfolding of the HFE protein. This in turn results in impaired expression of hepcidin, which controls the degradation of the membrane protein ferroportin on the enterocytes.
As a result, increased intestinal iron absorption leads to systemic iron overload in several organs, which can subsequently cause liver cirrhosis, cardiomyopathy and diabetes mellitus (bronchial diabetes). One of the most important complications of hereditary haemochromatosis is hepatocellular carcinoma. If liver damage of a different origin occurs at the same time, the risk of chronic liver damage with fibrosis is significantly increased.
Due to its slow and asymptomatic course over a long period of time, hereditary haemochromatosis often only manifests itself between the ages of 40 and 60, particularly in men, but not all carriers of this mutation necessarily develop the disease (incomplete penetrance).
The treatment of hereditary haemochromatosis consists of lifelong phlebotomy to normalize the iron concentration in the body. If this therapy begins before the manifestation of liver cirrhosis, organ damage can be averted in many patients. Chelating agents such as deferoxamine can be used as medication, but these have a high potential for side effects.
The development of CRISPR/Cas technology represents a milestone in biotechnological research. The molecular gene scissors make it possible to create precise DNA double-strand breaks in the genome of a cell. The subsequent, far less precise repair by special cellular enzymes, also known as non-homologous end joining, then leads to an interruption of the respective gene sequence, which makes the classic CRISPR/Cas technology a useful tool for gene silencing in the laboratory.
However, the correction of a point mutation or even the precise insertion of a specific gene sequence into the genome is much more difficult. In contrast to classic CRISPR/Cas, base editing no longer results in a DNA double-strand break, but in the targeted replacement of a single nucleotide - with high efficiency.
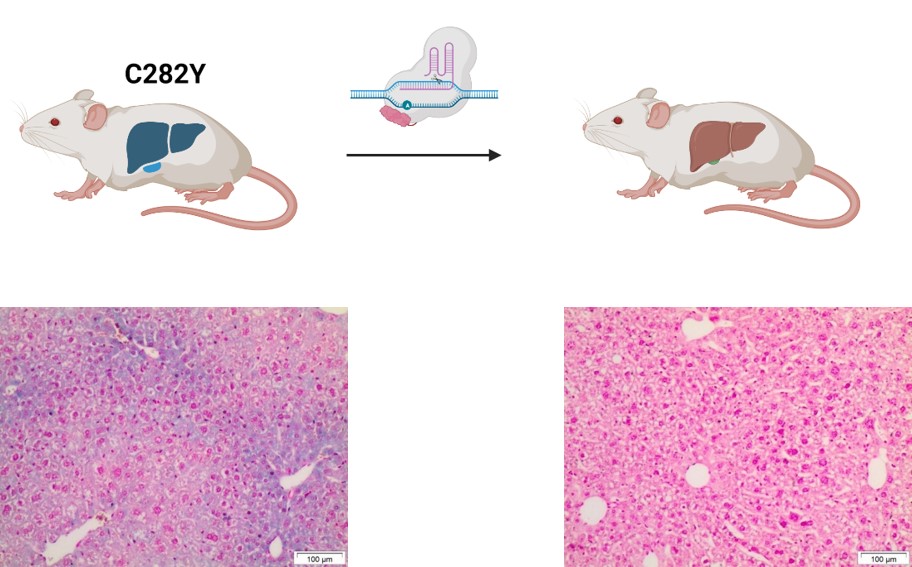
Using such a novel base editor, we have succeeded in correcting the C282Y mutation in vivo in a hemochromatosis mouse model. First, the base editor was configured to find and efficiently edit the correct "address" in the genome. Adeno-associated viral (AAV) vectors were then used to introduce the base editor into the hepatocytes. Significant improvements in iron parameters were observed after a single application.
Using the example of haemochromatosis, we were able to demonstrate efficient in vivo correction of the most common mutation in Europe. We are currently working on the use of non-viral vectors in order to provide an additional therapy option for severe courses of this and other metabolic diseases in the future.
For further information, please contact Dr. Simon Krooss(krooss.simon@mh-hannover.de, 0511-532 7128) and Prof. Dr. Michael Ott(Ott.Michael@mh-hannover.de, 0511-532 7120).
The original paper with first author Dr. Alice Rovai was published in the renowned journal Nature Communications .